The global antibiotic pipeline is in bad shape. Bacterial infections that were reliably treatable 30 years ago now kill people, and new antibiotic approvals have been slow. Natural products - compounds produced by bacteria, fungi, and plants - have historically been the best source of new ones. Penicillin, vancomycin, erythromycin: all came from organisms that had already figured out how to kill bacteria.
The problem is there are a lot of natural products to look through. The COCONUT database alone has 715,822 of them.
In this case study, K-Dense Web processed that entire database to produce 50 prioritized antimicrobial candidates ready for experimental screening. The whole pipeline ran in about 45 minutes.
Finding needles in a molecular haystack
Manually evaluating 715,000+ compounds isn't realistic. You need a way to filter, cluster, and prioritize before anything goes near a lab.
K-Dense Web was given a single prompt describing the research goal, then designed and ran the rest.
The pipeline
Step 1: Data preparation
K-Dense Web downloaded the full COCONUT database (664 MB), filtered for bacterial-derived compounds, and validated all SMILES structures using RDKit.
- Compounds downloaded: 715,822
- Bacterial-derived compounds: 24,911 (3.48%)
- Validation rate: 99.996%
- Final dataset: 24,910 unique compounds with validated structures
Step 2: Feature engineering
For each compound, K-Dense Web calculated physicochemical properties (molecular weight, LogP, TPSA, hydrogen bond donors/acceptors), structural descriptors (ring count, aromatic rings, fraction sp3 carbons), and drug-likeness metrics (QED score, Lipinski's Rule of 5 compliance, PAINS filtering).
| Property | Mean | Range |
|---|---|---|
| Molecular Weight | 539 Da | 1 - 4,900 Da |
| LogP | 2.1 | -29 to 37 |
| QED Score | 0.36 | 0.01 - 0.94 |
| Lipinski Compliant | 44.8% | - |
Only 39.7% of compounds passed both Lipinski's Rule of 5 and PAINS filters. That's not surprising - bacterial natural products often sit outside traditional drug-like chemical space. Vancomycin and daptomycin wouldn't pass Lipinski either.
Step 3: Chemical space analysis
The original plan was to pull bioactivity training data from ChEMBL and build a supervised model. The ChEMBL API returned errors. Rather than stopping, K-Dense Web switched to an unsupervised approach - which worked fine.
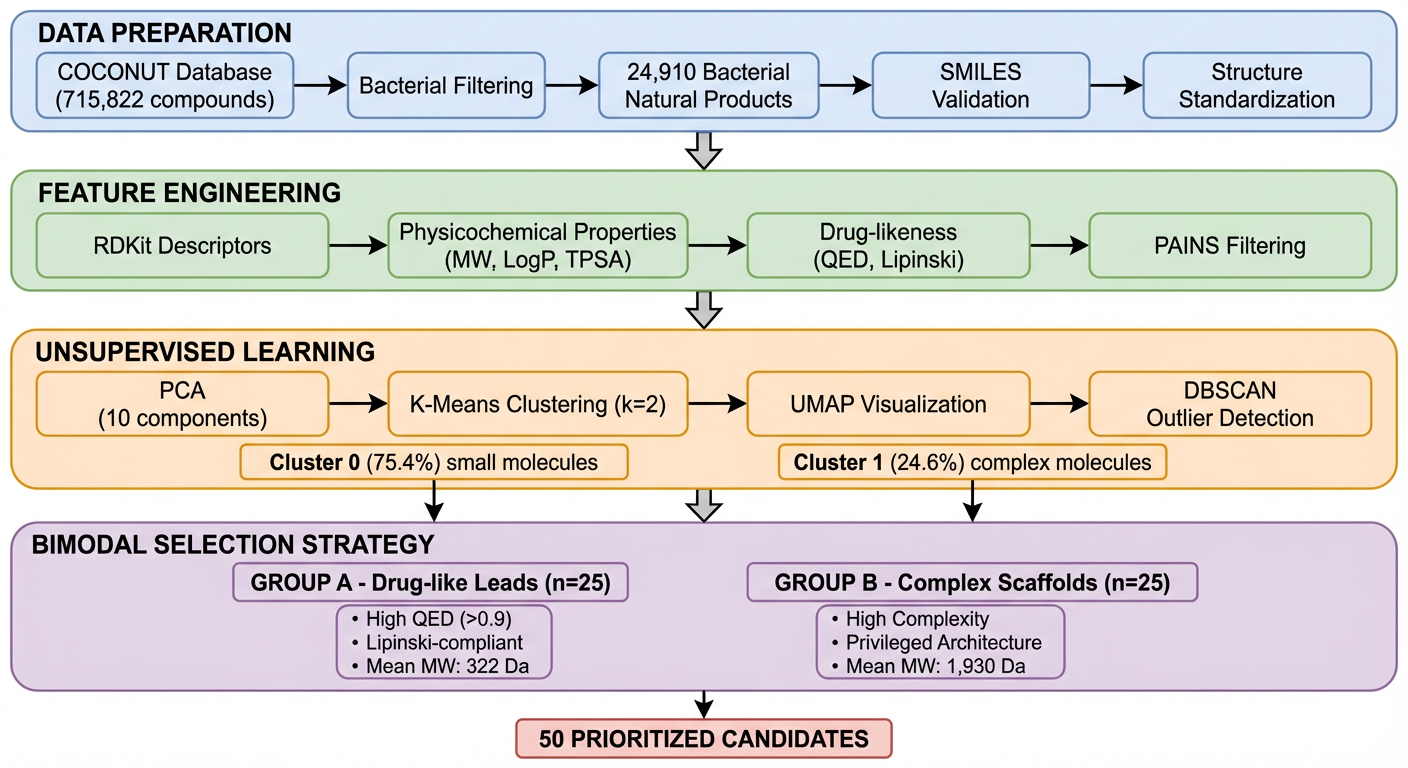
PCA of the full compound set turned up two distinct clusters:
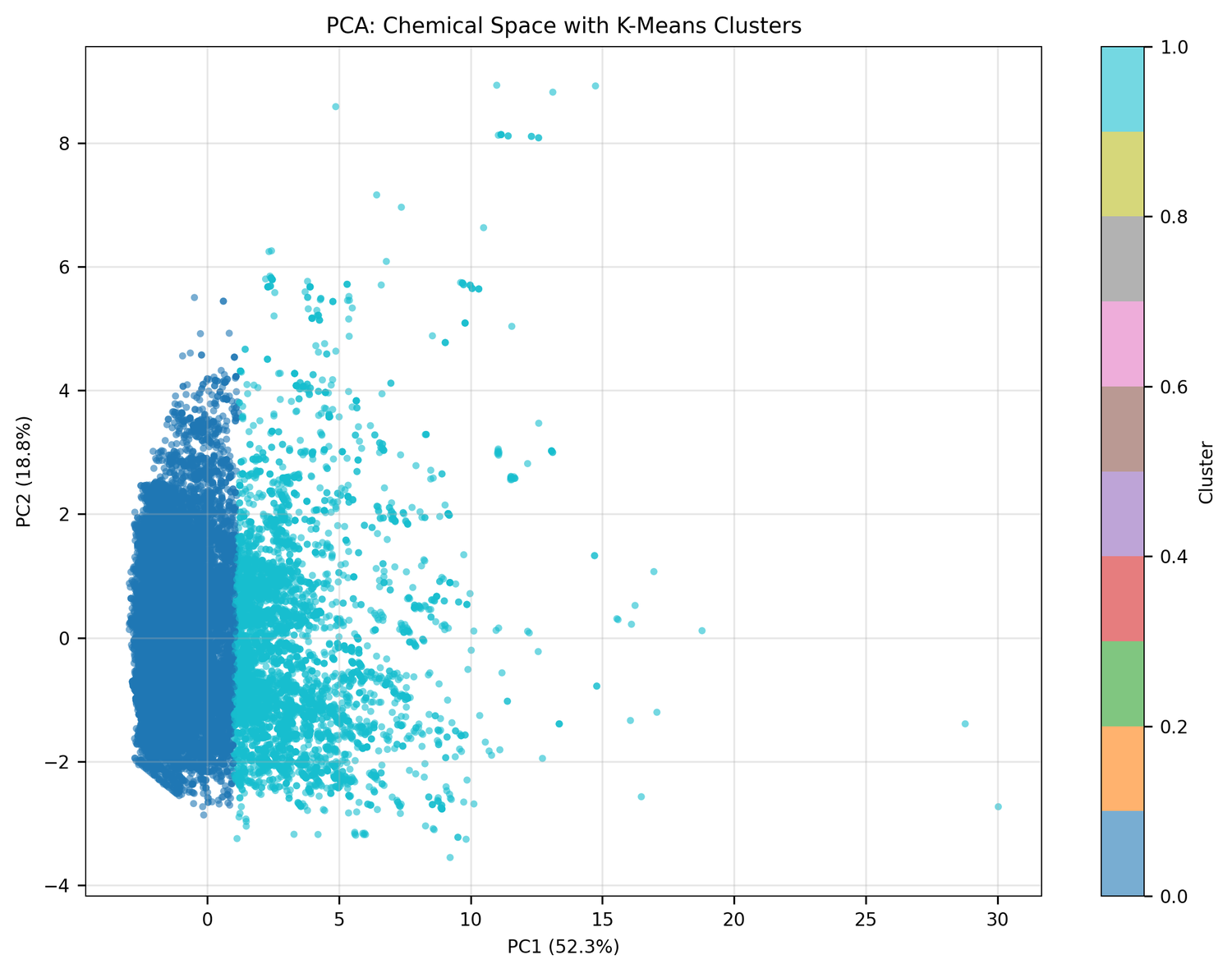
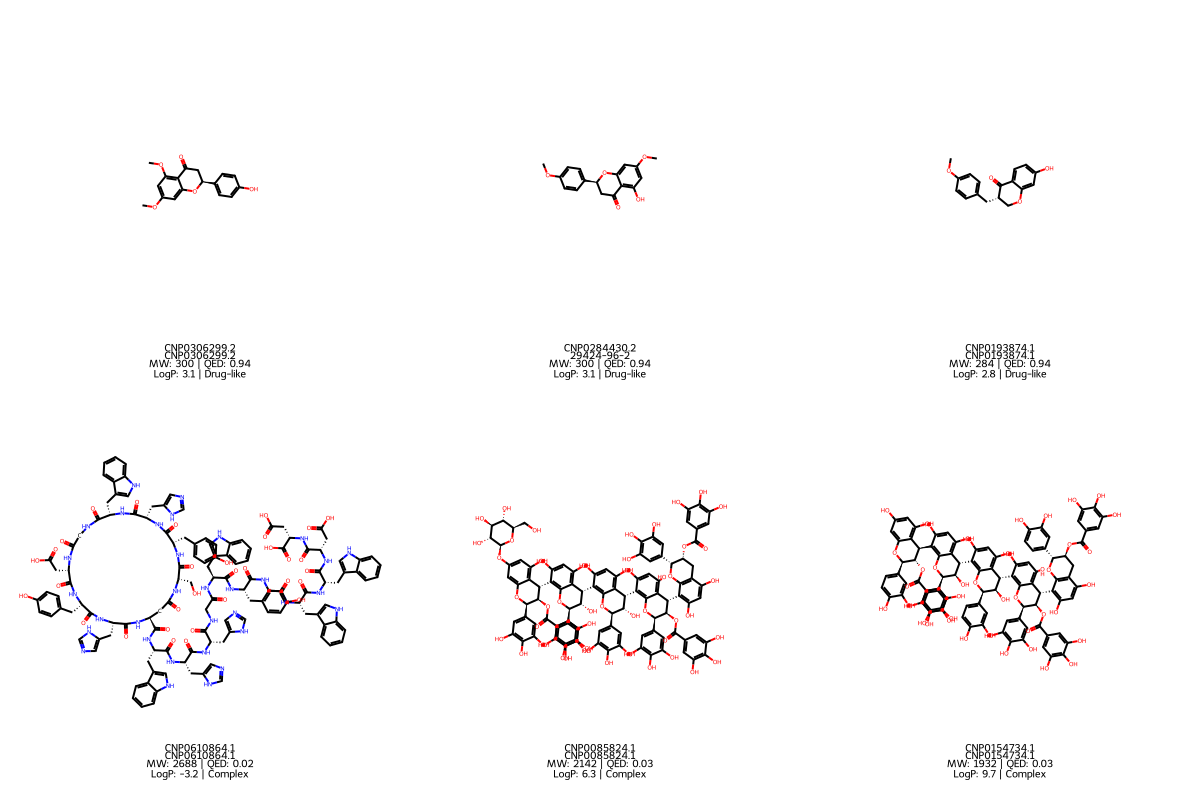
Cluster 0 (75.4% of compounds): Small, drug-like molecules. Mean MW: 396 Da, mean QED: 0.44. Probably alkaloids, terpenoids, and smaller polyketides.
Cluster 1 (24.6% of compounds): Large, complex molecules. Mean MW: 978 Da - 2.5× larger - mean QED: 0.09. Probably glycopeptides, lipopeptides, and macrocyclic antibiotics.
This bimodal split maps cleanly onto what's already known about antimicrobial natural products: one population of small, simple molecules and another of the kind of complex scaffolds that produced vancomycin.
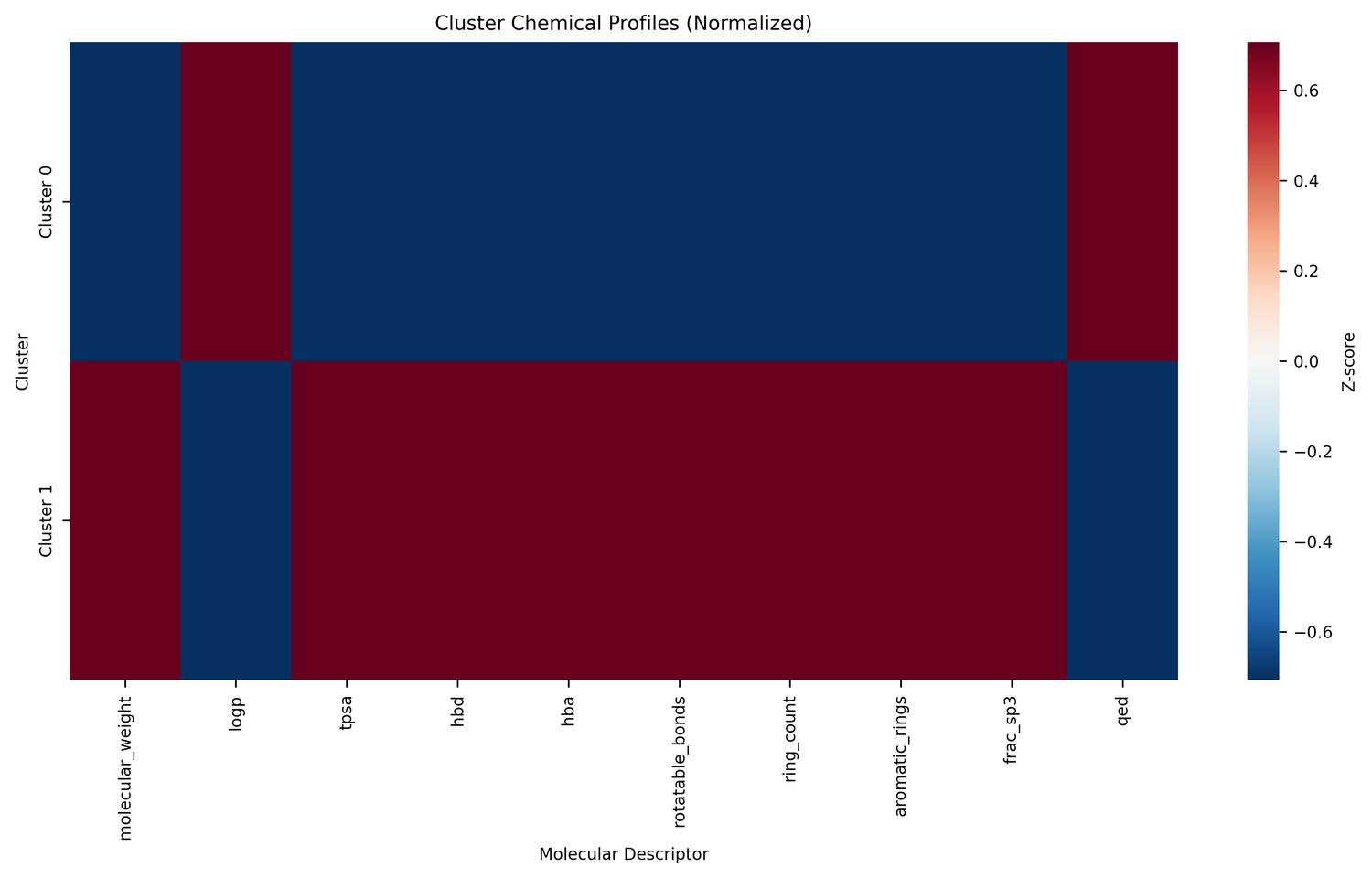
Step 4: Candidate selection
K-Dense Web selected 25 compounds from each cluster:
Group A: Drug-like leads (from Cluster 0)
- Mean MW: 322 Da, mean QED: 0.93
- 100% Lipinski compliant
- Good starting points for traditional medicinal chemistry optimization
Group B: Complex scaffolds (from Cluster 1)
- Mean MW: 1,930 Da, mean QED: 0.05
- Structural types typical of clinically successful antibiotics
- Lower development tractability, higher novelty potential
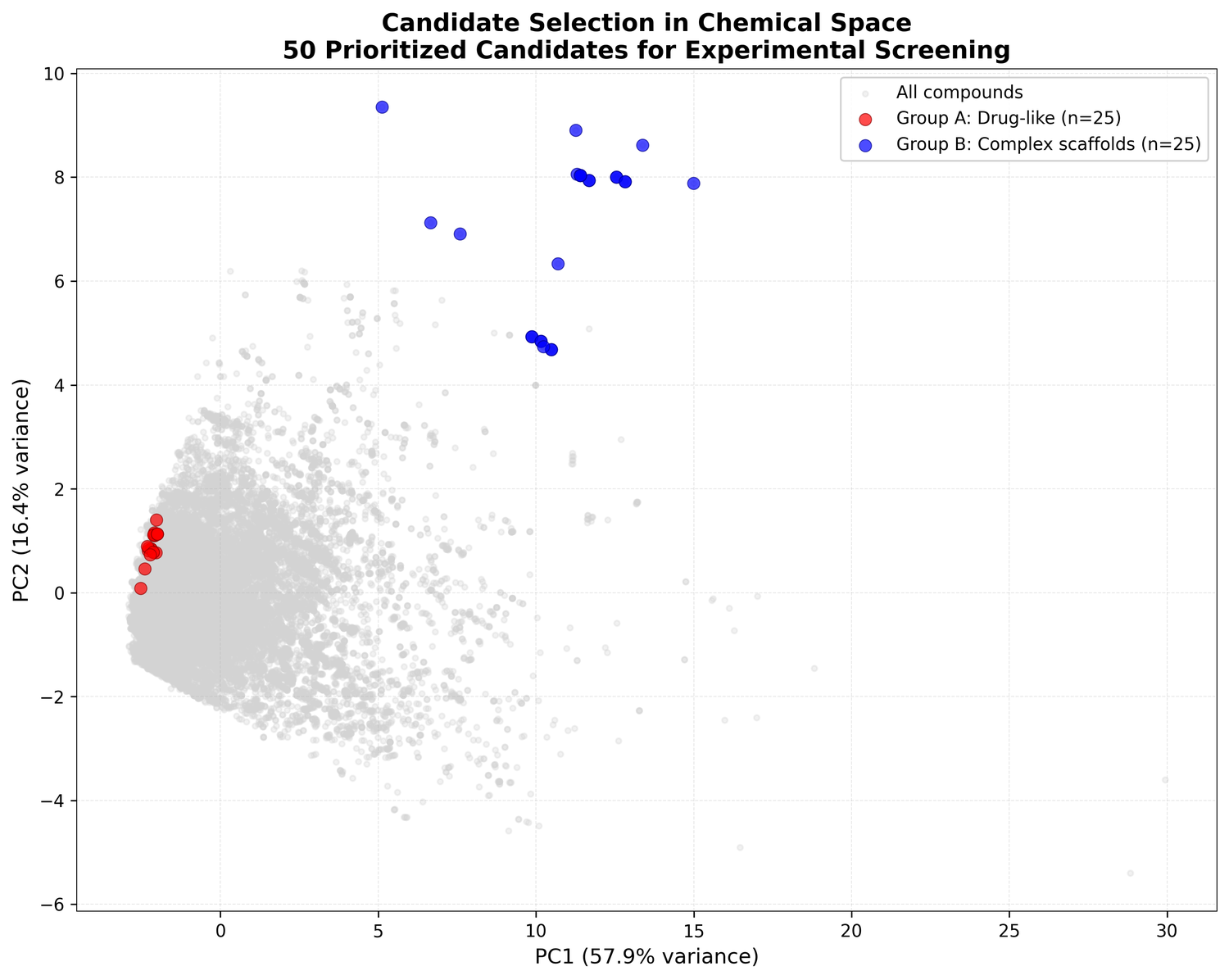
Covering both groups hedges the bets. Group A is easier to develop and optimize. Group B is where the bigger swings are.
Step 5: Validation and reporting
K-Dense Web produced 10 publication-ready figures, a research manuscript with methods, results, and discussion sections, and detailed candidate profiles.
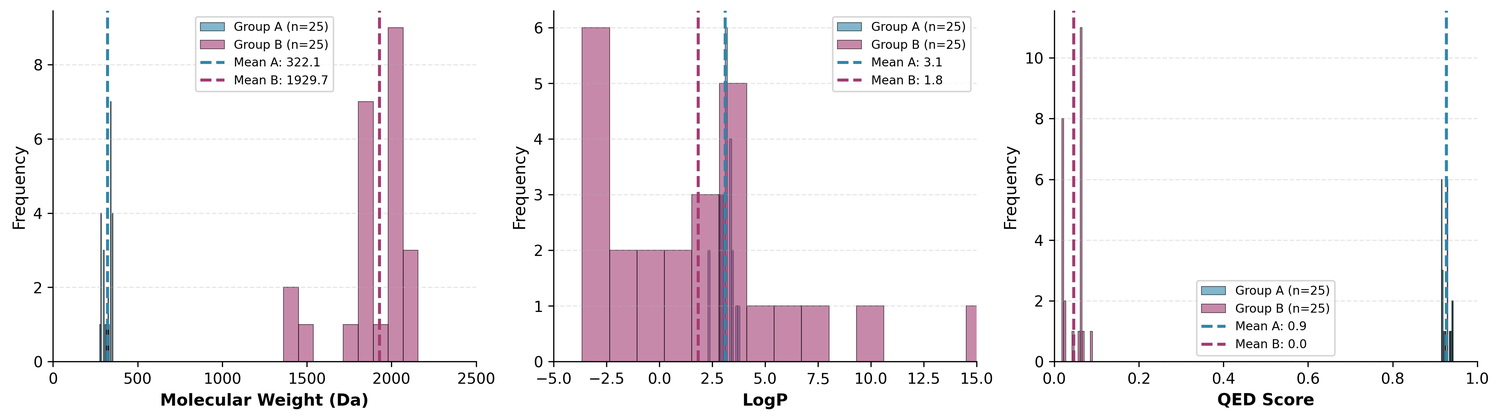
The two groups look quite different from each other:
Results
| Metric | Value |
|---|---|
| Initial compounds screened | 715,822 |
| Bacterial compounds identified | 24,910 |
| Chemical clusters discovered | 2 |
| Prioritized candidates | 50 |
| Group A (drug-like) | 25 |
| Group B (complex) | 25 |
| Pipeline execution time | ~45 minutes |
What this workflow replaced
Traditional computational drug discovery involves real setup time: installing RDKit, figuring out ChEMBL's API, writing clustering code, debugging visualization scripts. When something breaks mid-analysis - like an API going down - you lose time replanning.
K-Dense Web ran the full pipeline, handled the API failure without stopping, and produced figures and a manuscript draft at the end. The 45-minute runtime is mostly compute, not planning or debugging.
The 50 candidates are ready for antimicrobial screening against resistant strains (MRSA, VRE, MDR pathogens), MIC determination, and structure-activity relationship analysis using the chemical space clusters.
Try it yourself
Start your autonomous research project on K-Dense Web →
This case study was generated from K-Dense Web. View the complete example session including all analysis code, data files, figures, and the publication-ready research manuscript.